WordPress is too sluggish: it took >20 seconds to get this editor window to load (slow Chinese internet, but still). Since Substack is now also doing RSS & all the other stuff, I think it’s a good platform and I am moving there.
This post describes grit, which is an vaporware alternative interface to git (grit does not exist).
There are a few alternative interfaces to git, but they typically make the same mistake: they reason that git is complex because it supports many workflows that are only rarely useful, while 10% of the functionality would fill the needs of 90% of the uses. Git’s separation of mechanism and policy is confusing. They then conclude that, therefore, a more opinionated tool with built-in support for a small number of common workflows would be better.
I think this throws out the baby with the bathwater: git’s flexibility is its strength, its problems are rather that the user interface is awful. These are conceptually simpler problems. If you build a less powerful version, this also ensure that the system is not fully compatible with git and, therefore, I cannot recommend it as “better git” to students
Here, I describe an alternative, in the form of an imaginary tool, called grit (named for the most important quality in a student learning git). Grit could be just an alternative interface to a git repository that is completely, 100% compatible, enabling everything that git already does. In fact, it adds one important piece of extra complexity to git’s model.
Here is what is (a part of what) wrong with git and how grit fixes them:
1. The git subcommand names are a mess
There has been progress on this front with the introduction of git switch and friends, which fixes the worse offender (the overloading of git checkout to mean a million different operations), but it’s still a mess.
Grit uses a multi-subcommand interface. For example:
grit branch create
grit branch list
grit branch delete and, if there are unmerged commits: grit branch delete --force
grit branch rename
and so on… In particular, grit branch prints a help message.
Deleting a remote branch in grit is grit branch delete-remote origin/mytopic (as opposed to git push origin :mytopic — seriously, who can ever discover that interface?). Can you guess what grit tag delete-remote does?
Git has the concept of porcelain vs. plumbing, which is actually a pretty great concept, but packaged very badly. This is the whole theme of this post: git-the-concept is very good, git-the-command-line-tool is very bad (or, using the convention that normal font git refers to the concept, while monospaced git refers to the tool: git is good, but git is bad).
In grit, all the plumbing commands are a subcommand of internal:
grit internal rev-list
grit internal ls-remote
…
Everything is still there, but casual users clearly see that this is not for them.
2. Grit has a grit undo subcommand
Not only is the perhaps the number one issue that people ask about git, it is absurd that it is so for a version control system! Fast and easy undo is a major selling point of version control, but with git, undoing an action takes some magical combination of git reflog/git reset and despair. In grit, it’s:
grit undo
That’s it! If the working space has not been changed since the last grit command, then it brings it (and internal git status) back to where it was before you used the command. grit anything-at-all && grit undo is always a no-op.
This requires a new concept (similar to stash) in git to store the undo history, but is such an obvious sore point that it’s worth the extra complexity.
Technicalities: Using the option --no-undo means that the command should not generate an undo history entry. While keeping track of undo history is often cheap, there are a few exceptions. For example, grit branch delete-remote requires one to fetch the remote branch to be able to undo its deletion later and --no-undo skips that step for speed. If commits would be lost, the user is prompted for confirmation (unless --force is used, in which case, the branch is deleted, promptly, forcefully, and forever).
3. Grit has no multi-command wizards by default
Wizards are typically associated with graphical interfaces: a series of of menus where the user inputs all the information needed for a complex task.
Amazon’s checkout wizard is one many of us use regularly, but here is an example from the Wikipedia page on Wizards:
Kubuntu install wizard
On the command-line there are two possibilities of how to build wizards: (i) you open a command line dialog (do you want to continue? [Y/n] and so on) or (ii) you require multiple command invocations and keep state between them. Regular git has both of these, but it prefers to use (ii), which is the most complicated.
For an example of (ii): if you rebase and there is a conflict, it will drop you into the shell, expect you to fix the conflict, require a few git add operations, then git rebase --continue. Many git commands take --continue, which is a sure sign of a wizard.
For an example of (i), you can use git add -p: it will query you on the different chunks, and at the end execute the operation. Git add -p is actually great in that even if you have already accepted some chunks, if you use CTRL-C to quit, it will cancel the whole operation.
grit also has both, but prefers to use (i) whenever possible. If there is a conflict, it will start an interface similar to the existing git add -p and work chunk-by-chunk. It can start a subshell if you need more time or leave the state in a suspended animation (like happens now with git), but that is not the default. If you abort the operation (CTRL-C, for example), it cancels everything and leaves you in same situation as before you started the operation.
Arguably, the use of the index (staging area) can be seen as a form of a having a commit wizard, but it’s so fundamental to git that grit keeps it.
4. Grit performs no destructive actions without the --force flag!
With git, it’s impossible to know whether a command will destroy anything. For example, when merging, it may or may not work:
If there is a conflict, it will clobber your files with those awful <<<<< lines!
This happens with a lot of git commands: git checkout may or may not overwrite your changes (causing them to be thrown away). If fact, anything that causes a merge may lead to the conflict situation.
With grit, if something is non-trivial to merge or will potentially destroy an existing file, it will either (1) refuse to do it or require confirmation or (2) open a wizard immediately. For example, merge conflicts result in a wizard being called to merge them. If you want the old-school clobbering, you can always choose the --conflict-to-file option (on the command line or in the wizard itself).
Final words: grit does not exist, so we cannot know whether git’s problems really are mostly at the surface or if a deeper redesign really is necessary. Maybe something like grit will be implemented, and it will turn out that it is still a usability nightmare. However, one needs to square the git circle: how did it win the version control wars when it so confusing? It was not on price and, particularly for the open-source world, it was not by management imposition. My answer remains that git’s power and flexibility (that derives from its model as a very flexible enhanced quasi-filesystem under the hood) are a strength that it worth knowing about and climbing that learning curve, but git’s command line interface is an atrocious mess of non-design.
https://en.wikipedia.org/wiki/Naming_taboo “The naming taboo of the state (国讳; 國諱) discouraged the use of the emperor’s given name and those of his ancestors… The custom of naming taboo had a built-in contradiction: without knowing what the emperors’ names were, one could hardly be expected to avoid them”
Haskell rewritten to look like a “standard programming language”: Hinc . What is interesting is that makes you think how much of one’s impressions of programming languages is based on superficial features.
One of the strangest axioms to persist is that companies are designed for short-term thinking, unsuitable for deep work. Meanwhile, academics are chasing grants every year. Feels like a myth that discourages talented people from pursuing work in the private sector. https://t.co/qncDU4d5Fs
I have recently switched my thinking to embrace remote as the status quo rather than a short-term situation and as part of that move, we are accepting remote interns for Fall 2020.
Thus, we are extending the scheme and accepting remote interns. There are many possible projects and we will try to design a project to fit the student, but they can be either focused more on algorithms and tools or on biological problems.
We are going to have an Open Office Hours (i.e., a Zoom call where you can ask about anything, but this time focused on the remote internship options) on September 9 2020 @ 11am UTC (check your timezone!). Email me for the invite link.
As background, in our group, we have made a public commitment to supporting tools over at least 5-years, starting with the date of publication.¹
I have increasingly become convinced that code made available for reproducibility, what I call Extended Methods, is one thing that should be encouraged and rewarded, but is very different from making tools, which is another thing that should be encouraged and rewarded. The criteria for evaluating these two modes of code release should be very different. The failure to distinguish between Extended Methods code and tools plagues the whole discussion in the discipline. For example, internal paths may be acceptable for Extended Methods, but should be an immediate Nope for tools. I would consider this as immediate cause to recommend rejection if a manuscript was trying to sell code with internal path as a tool. On the other hand, it should also be more accepted to present code that is explicitly marked as Extended Methods and not necessarily intended for widespread use. Too often, there is pressure to pretend otherwise and blur these concepts.
Our commitment is only for tools, which we advertise as for others to use. Code that is adjacent to a results-driven paper does not come with the same guarantees! How do you tell? If we made a bioconda release, it’s a tool.
Our long-term commitment has the most impact before publication! In fact, ideally, it should have almost no impact on what we do after publication. This may sound like a paradox, but if tools are written by students/postdocs who will move on, the lab as a whole needs to avoid being in a situation where, after the student has moved on, someone else (including me!) is forced to debug unreadable, untested, code that is written in 5 different programming languages with little documentation. On the other hand, if we have it set up so that it runs on a continuous integration (CI) system with an exhaustive test suite and it turns out that a new version of Numpy breaks one of the tests, it is not so difficult to find the offending function call, add a fix, check with CI that it builds and passes the tests, and push out a new release. Similarly, having bioconda releases, cuts down a lot of support requests to use bioconda to install.
¹ In practice, this will often be a longer period, on both ends, as tools are supported pre-publication (e.g., macrel is only preprinted, but we will support it) and post-publication (I am still fixing the occasional issue in mahotas, even though the paper came out in 2013).
As we try to move the Macrel preprint through the publishing process, one reviewer critique was likely caused by the reviewer using our webserver in the incorrect way. We support using either DNA contigs or peptides input, but it is technically possible to use it in a wrong way by feeding it peptides and telling it they’re DNA (or the other way around).
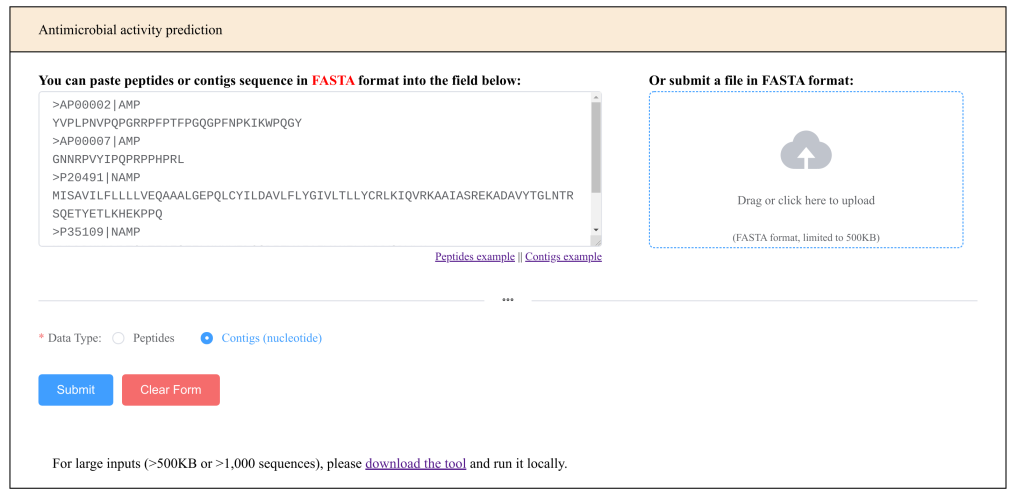
The easy response would have been “this was user error. Please do not make mistakes.” Instead, we looked at the user interface and asked if this was instead a user interface bug. This is what it used to look like:
Advantages:
It has a big text box where you input: this is pretty obvious what to do
We accept a file upload too
The Data Type field has a red star indicating it’s mandatory. This is a pretty typical convention. Hopefully, many users will recognize it
It is still quite easy, however to make mistakes:
See how the user selected “Contigs (nucleotide)”, but the sequences are clearly amino-acids. The phrasing is not so clear. Having two ways to execute the service (a textbox and an upload box) was also potentially confusing: if you ask me now what happens if you type in something and upload a file, the answer is that I don’t know.
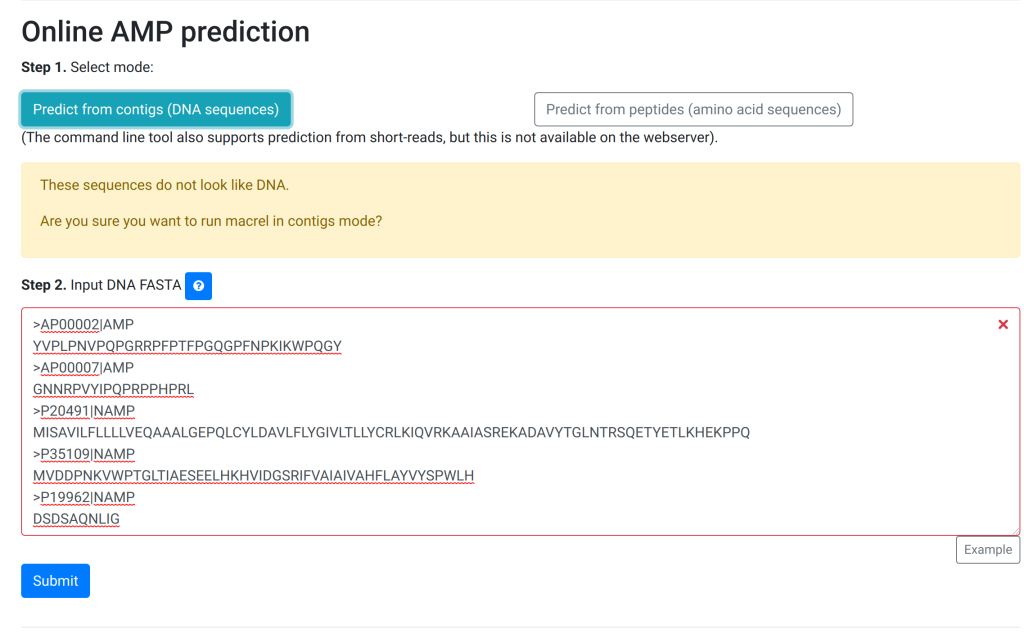
New Version
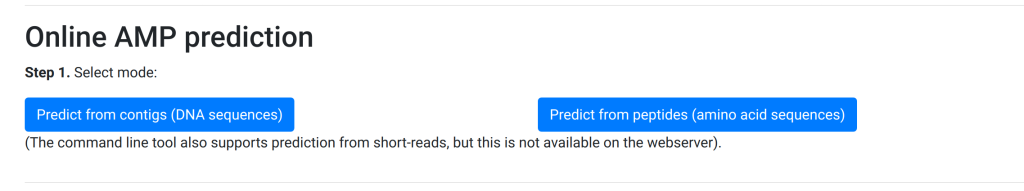
In the new version (currently online), we rejigged the process:
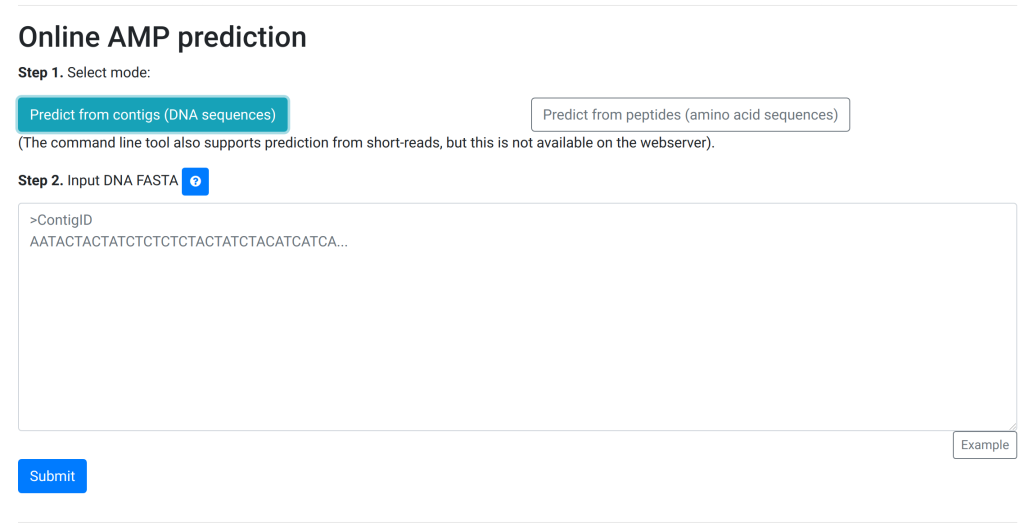
The user first has to explicitly select the mode and only then can they progress and see the textbox:
Now, the guidance is also explicit “Input DNA FASTA”. More importantly, if they make a mistake, we added some checks:
Advantages:
More explicit choice for the user, so less room for careless mistakes. Also, the instruction text adapts to the previous choice as does the filler text.
Error checking on the fly
Simpler: only a big text box, no uploading of a FASTA file.
The last one can sound like “bad usability”: the tendency in some usability circles to privilege first-time and casual users over power-users. Also, having to click on one button to get the textbox may similarly be better for first-time users, while ultimately annoying for power-users (although this is all client-side, so it’s very fast).
However, I argue that, in this case, power-users are not our main concern: Power users should be using the command-line tool and that will enable them to overcome any limitations of the website. We also put in work into the usability of the command-line tool (which is equally important: just because it’s a command line it doesn’t mean you do not need to care about its usability).
PS on implementation: I wrote the code for the frontend in elm, which was incredibly pleasant to work with elm even though it was literally the first time I was using the language. It is just nice to use a real language instead of some hacky XSLT-lookalike of the kind that seems popular in the JS world. The result is very smooth, completely running client-side (except for a single API call to a service that wraps running macrel on the command line).
We’ve started blogging a bit as a group on our website, which is a development I am happy about.
http://big-data-biology.org/blog/2020-04-29-NME/ : The famous Tank story of a neural network that purported to tell apart American from Soviet tanks, but just classified on the weather (or time of day) is probably an urban legend, but this one is true. What I like about this type of blogging is that this is the type of story that takes a lot of time to unravel, but does not make it to the final manuscript and gets lost. [same story as Twitter thread]. Most of the credit for this post should go to Célio.
http://big-data-biology.org/blog/2020-04-10-cryptic : A different type of blogpost: this is a work-in-progress report on some our work. We are still far away from being able to turn this story into a manuscript, but, in the meanwhile, putting it all in writing and out there may accelerate things too. Here, Amy and Célio share most of the credit.
2. We’ve updated the macrel preprint, which includes a few novel things. We have also submitted it to a journal now, so fingers crossed! In parallel, macrel has been updated to version 0.4, which is now the version described in the manuscript.
3. NGLess is now at version 1.1.1. This is mostly a bugfix release compared to v1.1.0.
4. We submitted two abstracts to ISMB 2020: The first one focuses on macrel, while the second one is something we should soon blog about: we have been looking into the problem of predict small ORFs (smORFs) more broadly. For this we took the dataset from (Sberro et al., 2019) which had used conservation signatures to identify real smORFs in silico and treated it as a classification problem: is is possible to based solely on the sequence (including the upstream sequence) to identify whether a smORF is conserved or not (which we are taking as a proxy for it being a functional molecule).
This is not a real question, we know that yes, unfortunately, they did. There are over one thousand confirmed cases. But a recent preprint, posits that over 40,000 and maybe as many as 80,000 people did.
The preprint is not very informative on methods, but, it seems to me, that, if this was the only evidence we had about Santa Clara County, it would barely be enough, according to the traditional standards of scientific evidence, for the authors to claim that anyone in Santa Clara got infected. In any case, the prevalence reported is likely an over estimate.
The evidence
The authors tested 3,330 individuals using a serological (antibody) test and obtained 50 positive results (98.5% tested negative, 1.5% tested positive).
This test was validated by testing on 401 samples from individuals who were known to not have had covid19. Of these 401 known negatives, the test returned positive for 2 of them. The test was also performed on 197 known positive samples, and returned positive for 178 of them.
There are some further adjustments to the results to match demographics, but they are not relevant here.
The null hypothesis
The null hypothesis is that none of the 3,330 individuals had ever come into contact with covid19 and had no antibodies.
Does the evidence reject the null hypothesis?
Maybe, but it’s not so clear and we need to bring in the big guns to get that result. In any case, the estimate of 1.5% is almost certainly an over-estimate.
Naïve version
Let’s estimate the specificity: the point estimate is 99.5% (399/401), but with a confidence interval of [98.3-99.9%]
With the point estimate of 99.5%, then the the p-value of having at least one infection is a healthy 2·10⁻¹¹
However, if the confidence interval is [98.3-99.9%], we should perhaps assume a worst-case. If specificity is 98.3% then the number of positive tests is actually slightly lower than expected (we expected 57 just by chance!). Only if the specificity is above 98.7% do we get some evidence that there may have been at least one infected person.
With this naive approach, the authors have not shown that they were able to detect any infection.
Semi-Bayesian approach
A more complex model samples over the possible values of the specificity
If the specificity is modeled as a random variable, then we have observed 2 positives out of 401 in known cases.
So, we can set a posterior for it of Beta(402, 3) (assuming the classical Beta(1,1) prior).
Let’s now simulate 3330 tests, assuming they are all negative, with the given specificity.
If we repeat this many times, then, about 7% of the times, we get 50 or more false positives! So, p-value=0.07. As the olds say, trending towards significance.
Still no evidence for any infections in Santa Clara County.
Full-Bayesian
Finally, the big guns get the right result (we actually know that there are infections in Santa Clara County).
We now do a full Bayesian model
We model the specificity as above (with a prior Beta(1,1)), but keep also model the true prevalence as another Bayesian variable.
Now, we have some true positives in the set, given by the prevalence.
We model the sensitivity in the same way that we had modeled the specificity already, as a random variable constrained by observed data.
We know that the true + false positives equals 50.
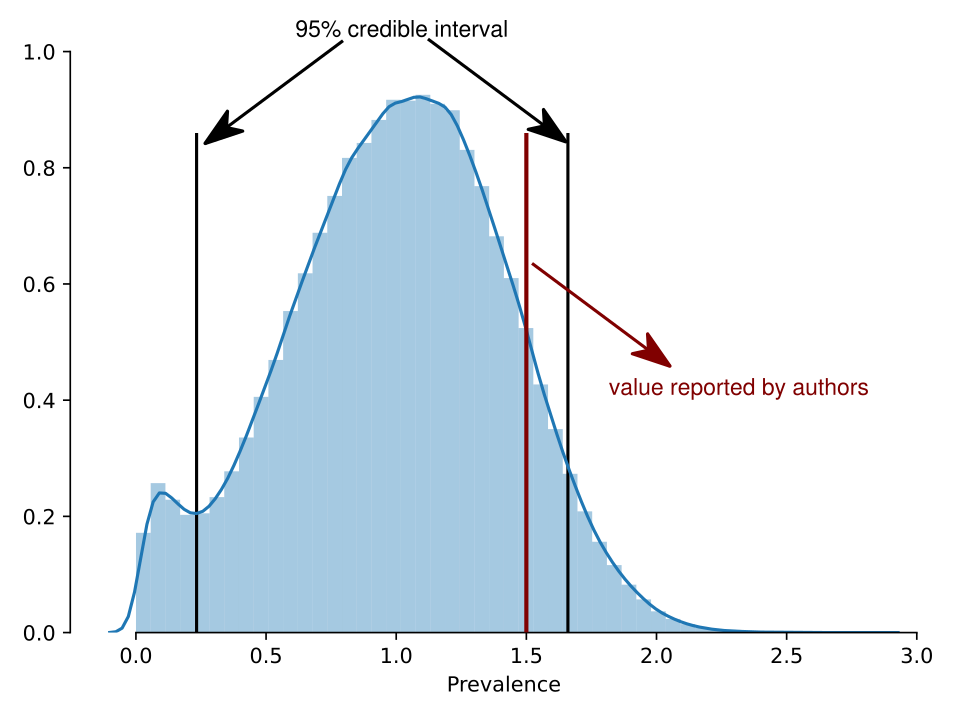
Now the prevalence credible interval is (0.4-1.7%). It is not credible that there are zero individuals who are positives anymore. The credible interval for the number of positives in the set is (15-51).
The posterior distribution for the prevalence is the following:
In this post, I did not even consider the fact that the sampling was heavily biased (it is: the authors recruited through Facebook, it can hardly be expected that people who are looking to get tested would not be enriched for those who suspect were sick).
In the end, I conclude that there is evidence that the prevalence is greater than zero, but likely not as large as the authors claim.