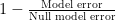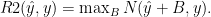An important component of my recent paper (previous post) on imaging protist (micro-eukaryotes) communities is a classifier that classifies each individual object into one of 155 classes. These classes are organized hierarchically, so that the first level corresponds to living/non-living object; then, if living, classifies it into phyla, and so on. This is the graphical representation we have in the paper:

Using a large training set (>18,000), we built a classifier capable of classifying objects into one these 155 classes with >82%.
What is the ML architecture we use? In the end, we use the traditional system: we compute many features and use a random forest trained on the full 155 classes. Why a random forest?
A random forest should be the first thing you try on a supervised classification problem (and perhaps also the last, lest you overfit). I did spent a few weeks trying different variations on this idea and none of them beat this simplest possible system. Random forests are also very fast to train (especially if you have a machine with many cores, as each tree can be learned independently).
As usual, the features were where the real work went. A reviewer astutely asked whether we really needed so many features (we compute 480 of them). The answer is yes. Even when selecting just the best features (which we wouldn’t know apriori, but let’s assume we had an oracle), it seems that we really do need a lot of features:
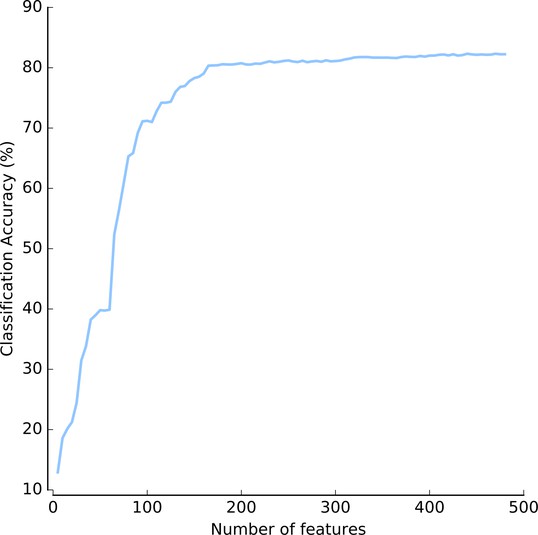
(This is Figure 3 — supplement 4: https://elifesciences.org/articles/26066/figures#fig3s4sdata1)
We need at least 200 features and it never really saturates. Furthermore, features are computed in groups (Haralick features, Zernike features, …), so we would not gain much
In terms of implementation, features were computed with mahotas (paper) and machine learning was done with scikit-learn (paper).
§
What about Deep Learning? Could we have used CNNs? Maybe, maybe not. We have a fair amount of data (>18,000 labeled samples), but some of the classes are not as well represented (in the pie chart above, the width of the classes represents how many objects are in the training set). A priori, it’s not clear it would have helped much.
Also, we may already be at the edge of what’s possible. Accuracy above 80% is already similar to human performance (unlike some of the more traditional computer vision problems, where humans perform with almost no mistakes and computers had very high error rates prior to the neural network revolution).
 where ŷi is the cross-validated predictions for input i ) has some nice properties. However, it is meaningless as a number and it would be nice to normalize it.
where ŷi is the cross-validated predictions for input i ) has some nice properties. However, it is meaningless as a number and it would be nice to normalize it.